



СИНДРОМ ОХТАХАРА/OHTAHARA SYNDROME/EARLY INFANTILE DEVELOPMENT

& EPILEPTIC ENCEPHALOPATHY (EIDEE)

MKБ-10 G40.3

ПРЕВАЛЕНЦА – непозната.
ИНЦИДЕНЦА – синдромот Охтахара е забележан кај приближно 1/50 000 раѓања во
Велика Британија до 1/100 000 живородени деца во Јапонија.
ВОВЕД
Синдромот Охтахара е ретка форма на епилепсија која се карактеризира со напади и
доцнење во развојот кои обично се случуваат во првите три месеци од животот, но некои
се појавуваат во првите неколку недели по раѓањето (најчесто во првите 10 дена). Некои
случаи се предизвикани од мозочна абнормалност, метаболичко нарушување или генска
мутација, додека за други случаи не постои позната причина. Девојчињата и момчињата
се подеднакво погодени. Доенчињата најчесто имаат тонични напади (вкочанетост на
мускулите, нагорен поглед, проширени зеници и променето дишење), но можат да
доживеат и фокални напади (зафаќаат само една област или страна од мозокот) и ретко,
миоклонични напади (ненадејни грчеви или грчеви на горниот дел од телото, рацете и
нозете). Децата со нарушување можат да имаат повеќе од еден тип напади и симетрични
грчеви кои можат да се појават во групи или поединечно и можат да траат до 10 секунди.
ЕТИОЛОГИЈА
EIDEE може да биде резултат на различни етиологии, дури 80 % од случаите се поврзани
со структурни абнормалности на мозокот. Некои случаи се должат на метаболички
нарушувања (дефицит на цитохром C-оксидаза, дефицит на карнитин палмитоил
трансфераза II) или мозочни малформации (поренцефалија, хемигаленцефалија) кои
можат да бидат и генетски по потекло. Генетските варијанти на EIDEE се поврзани со
мутации во одредени гени, како што се ARX (Xp22.13), CDKL5 (Xp22), SL25A22 (11p15.5) и
STXBP1 (9q34.1), меѓу другите. Се смета дека генетските абнормалности водат до EIDEE
бидејќи се поврзани со невронска дисфункција или мозочна дисгенеза. Како
метаболитички причини можат да се вклучат и некетотичната хиперглицинемија,
нарушувањата на амино/органски киселини, нарушувањата на циклусот на уреа,
митохондријалните нарушувања, нарушувањата на пиридоксин и пиридоксал-5-фосфат,
недостатокот на сулфитна оксидаза, како и Менкесовиот синдром (Зелвегеров синдром).
Кај некои, EIDEE може да премине во Вестов синдром (помеѓу 2 до 6-месечна возраст), а
подоцна во синдромот Ленокс-Гасто. Одредени генетски варијанти се манифестираат со
дополнителни знаци, како што се дискинетични движења и атипичен фенотип на Ретов
синдром.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Новороденчињата со синдромот Охтахара развиваат чести напади во првите 3 месеци од
животот. Најчесто се појавуваат во првите 2 недели по раѓањето. Овие напади во некои
случаи тешко се контролираат. Тие често имаат потреба од лекови против напади.
Новороденчињата имаат слаби рефлекси за цицање, хипотонија и се манифестираат со
генерализирани напади. Моделот на овие грчеви останува непроменет за време на
будност и спиење, и тие можат да се појават по стопати на ден. Обично траат само
неколку секунди и можат да бидат во форма на вкочанетост на рацете и/или нозете,
влијаат на една или двете страни на телото, се јавуваат сами или во кластери, се јавуваат
кога бебето е будно или спие. Други типови напади, вклучувајќи генерализирани
тонично-клонични напади (напади кои влијаат на двете половини на мозокот и
започнуваат со ненадејно губење на будноста, проследено со мускулна вкочанетост и
грчеви движења), фокални моторни напади (можат да предизвикаат абнормални
движења на само еден дел од телото) и хемиконвулзии, се забележани во 1/3 од
случаите. Доенчињата со овој синдром имаат доцнење во развојот. Ова може да биде
резултат на основната причина за нивната состојба во комбинација со тековните напади.
Тие често не достигнуваат развој на иста возраст како и другите доенчиња. Тие исто така
можат да имаат абнормални резултати на невролошки преглед. Развојните симптоми
можат да бидат присутни уште пред да започнат нападите, кои можат да се влошат како
што се зголемува зачестеноста на нападите.
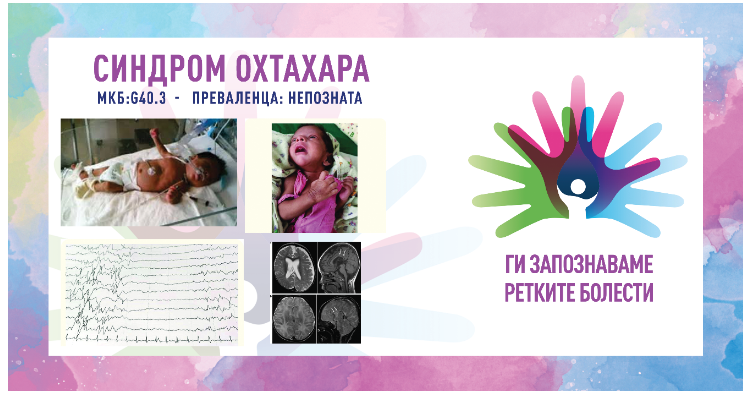
Дијагностицирањето се заснова на клинички и електроенцефалографски наоди.
Карактеристичниот електроенцефалограм (ЕЕГ) прикажува шаблон на потиснување на
пукање кој се појавува со почетокот на спазмите и кој се состои од рафали на шила со
висока амплитуда и полишици кои наизменично се менуваат со периоди на основен
ритам со низок напон (супресија). Оваа шема на ЕЕГ е континуирана и останува
непроменета и за време на будност и за време на спиење. Структурни абнормалности
често можат да се видат на кранијална МРИ. Кај некои новороденчиња се спроведуваат
лабораториски тестови за да се идентификува причината за синдромот, како и тестови за
урината насочени кон метаболитички нарушувања. Генетските тестови вклучуваат
хромозомски микронизи, генетски панели за епилепсија и секвенционирање на цели
егзоми кои можат да помогнат во пронаоѓање на причините за настанување на
синдромот Охтахара.
ПОСЛЕДИЦИ
Прогнозата е доста лоша, а смртта обично се јавува во детството (50 % пред возраст од 2
години). Преживеаните имаат тешки психомоторни оштетувања со континуирани
напади. Како што растат, кај некои деца се развиваат и други епилептични нарушувања
како што се синдромот Вест и синдромот Ленокс-Гесто. Членовите на семејството на
доенчињата можат да се консултираат со тим за палијативна нега бидејќи симптомите
можат да се влошат или да се развијат. Смртта често се должи на оптоварување од
напади, пневмонија или други компликации од моторни пречки.
ТРЕТМАН
Бидејќи не постои лек за EIDEE, пациентите имаат потреба од постојан надзор и грижа.
Третманот може да вклучува кортикостероиди, кетогена диета (со висока содржина на
масти, ниски јаглени хидрати) и физичка, говорна и професионална терапија.
Хирургијата на епилепсија, во случаи кога е присутна фокална мозочна лезија
(оштетување или абнормален развој на една област/страна од мозокот), може да биде
корисна затоа што антиепилептичните лекови, како што се бензодиазепините,
валпроатот, леветирацетам, зонисамид и фенобарбитал, покажале ограничен успех во
контролирањето на нападите, како и пиридоксинот. Забележано е дека кетогената диета
покажува одреден успех во контролата на нападите. Пациентите со EIDEE со одредени
структурни абнормалности имаат корист од хируршката интервенција, доколку е
еднострана. Повеќето случаи на EIDEE се спорадични („de novo“ мутации), па затоа се
препорачува генетско советување со цел да се информираат родителите дека нивниот
ризик да имаат понатамошни деца со EIDEE е низок.










{kind=link}
{kind=link}
{kind=link}
{kind=link}