



Во месецот со најмалку дена во годината, во месецот кој оваа година е редок по своите ретки 29 дена и во месецот кој пациентите со ретки болести го идентификуваат како СВОЈ, по шести пат ќе се обидеме поблиску да Ве запознаеме со ретките болести од кои заболува и со кои се соочува и живее населението од нашиот охридско – струшки регион. Да ги дознаеме предизвиците и проблемите со кои се соочуваат. Како ја добиваат дијагнозата? Дали има некаков лек? Колку и дали воопшто се пристапни и достапни лековите?…

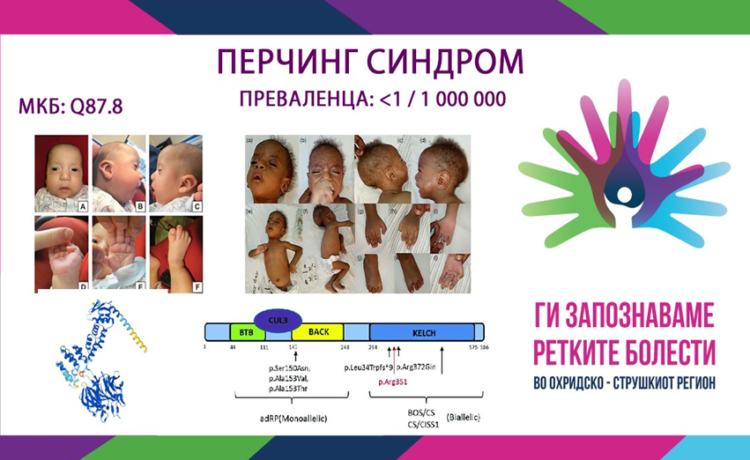
ПЕРЧИНГ СИНДРОМ/ PERCHING SYNDROME
MKБ-10 Q87.8
ПРЕВАЛЕНЦА – непозната, само 20 случаи се опишани во литературата.
ВОВЕД
Синдромот на Перчинг е автосомно-рецесивно мултисистемскo нарушување предизвикано од хомозиготни или сложени хетерозиготни патогени варијанти во генот KLHL7 (Kelch Like Family Member 7 (KLHL7)), кој се наоѓа на хромозомот 7p15 и кој се манифестира со глобален развоен застој, карактеристични црти на лицето, тешкотии со хранењето и дишењето, хипотонија и контрактури. Акронимот „PERCHING“ по кој е насловен синдромот се однесува на најкарактеристичните фенотипски елементи кои се манифестираат при истиот:
P – постурални и палатални абнормалности
E – егзофталмус и проблеми со зависност/хранење од ентерална цевка
R – респираторен дистрес и ретинитис пигментоза
C – контрактури и камптодактилија
H – хипертелоризам и хирзутизам
I – IUGR/дефицит во растот и интелектуална попреченост/задоцнување во развојот
N – nevus flammeus и невролошки малформации
G – лицево Гешталт/гримасирање и генитоуринарни абнормалности
Иако фенотипскиот спектар на овој синдром е широк и мултисистемски, постои специфична дисморфична презентација која може да го олесни клиничкото препознавање на овој синдром. Од првиот објавен случај за овој синдром во 2016 година (Angiuset 2016), до денес, во литературата се опишани само околу 20 пациенти.
ЕТИОЛОГИЈА
Мутациите во KLHL7 претходно се опишани како причина за констелација на клинички наоди со описи и на синдромот Cripponi (CS) – исто така познат и како Синдром на потење предизвикан од тип 1 (CISS1) и како презентација слична на БОС (Boring-Opitz синдром (BOS)). Афектираните деца имаат дизморфични карактеристики и доцнење во развојот и нивните клинички карактеристики не се преклопуваат доследно со фенотипски сличните CS/CISS1 и BOS. Исто така, афектираните браќа и сестри на опишаните случаи се разликуваат помеѓу себе што укажува на постоење на определена варијабилност во клиничката слика кај овој синдроом. Генот KLHL7 е лоциран на краткиот крак на хромозомот 7 и кодира протеин од семејството Келч. Протеините од семејството Келч играат важна улога во посредувањето на протеинските интеракции и затоа се важни за различни клеточни активности, вклучувајќи пролиферација, диференцијација, миграција, одржување на цитоскелетната структура и генска експресија. Прецизната функција на протеинскиот производ на KLHL7 останува нејасна, но познато е дека овој протеин е вклучен во препознавањето на супстратот како дел од комплексите на кулин-3 (CUL3) кои содржат Е3 убиквитин лигаза кои посредуваат деградација на протеозомот.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
Заедничките невроразвојни, мускулно-скелетни, дизморфни и други клинички манифестации на пациентите со биалелни мутации во генот KLHL7, дефинираат нова мултисистемска болест.
Дијагностицирањето на синдромот Перчинг вклучува низа чекори, како на пример:
- Клиничка евалуација
- Пред генетското тестирање, вообичаено се врши темелна клиничка евалуација од медицински генетичар и/или педијатар (детски невролог). Оваа евалуација може да вклучува преглед на личната и семејната историја на пациентот
- Собирање биолошки примероци од пациентот
- Биолошкиот примерок најчесто е крв или плунка
- Екстракција на ДНК
- Од примерокот се екстрахира ДНК
- Генетско тестирање може да вклучи разни методи за откривање на мутации во KLHL7
- Секвенционирање кое се спроведува според Sanger
- Секвенционирање со следната генерација технологии (NGS) кои овозможуваат симултано секвенционирање на повеќе гени, вклучувајќи го и KLHL7. Вообичаено се користат насочени генски панели или секвенционирање на цел егзом (WES).
- Арејната компаративна геномска хибридизација (aCGH) може да идентификува поголеми геномски промени, како што се делеции или дупликации во генот KLHL7
- Анализа на резултатите и интерпретација на генетските варијанти
- Добиените генетски резултати се анализираат со разни биоинформатички алатки кои овозможуваат споредба на ДНК-секвенцата добиена од пациентот со референтна секвенца, и пребарување на базите на податоци за вообичаената генетска варијабилност во популацијата, како и на базите податоци за генетска варијабилност поврзана со болести
- Толкување на варијантата
- Идентификуваните генетски варијанти се проценуваат за нивната потенцијална патогеност од страна на молекуларните генетичари, генетските советници и клиничките генетичари
Важно е да се напомене дека генетското тестирање во медицинската генетика треба да биде спроведено во соодветно акредитирана генетска лабораторија од страна на квалификувани генетичари или генетски советници. Дополнително, толкувањето на генетските варијанти може да биде сложено и често неопходна е консултација со специјалист по генетика за целосно да се разберат клиничките импликации на болеста и да се овозможи понатамошно планирање на семејството.
ПОСЛЕДИЦИ
Прогнозата зависи од тежината на тешкотиите со хранење и дишење, хипотонијата и од контрактурите.
ТРЕТМАН
Лекувањето на оваа болест е главно симптоматско и во зависност од клиничката манифестација често се јавува потреба од мултидисциплинарен пристап кој вклучува детски невролог, генетичар, ортопед, пулмолог, ендокринолог, офталмолог и/или детски хирург. За прогнозата на оваа болест не може да се каже многу со оглед на малиот број опишани пациенти и краткиот период на следење на природниот тек на болеста.
Генетско советување
Доколку се идентификува генетската причина за болеста кај пациентот, вообичаено на родителите им се нуди тестирање за носителство. Најчесто, двајцата родители се здрави носители за оваа автосомно-рецесивна болест, и во тој случај, веројатноста за повторување на болеста во која било следна бременост е 1 од 4 или 25 %. Генетската дијагноза отвора можности за пошироко каскадно тестирање за носителство во семејството и овозможува планирање на семејството во смисла на пренатална или предимплантациона генетска дијагностика.
за проектот:
Здружението на пациенти Ретко е да си редок од Охрид и овој февруари, во рамките на шестата сезона од проектот ГИ ЗАПОЗНАВАМЕ РЕТКИТЕ БОЛЕСТИ 2024, Ве запознаваат со ретките болести од кои заболува населението во охридско – струшкиот регион, со застапеност на истите низ различни градови во Р.С. Македонија, со надеж дека преку овие нови 29 ретки дијагнози сите заедно ќе успееме да го кренеме гласот за проблемите со кои се соочуваат лицата со ретки болести, но и нивните семејства.
Кампањата е од информативно – едукативен карактер и на неа работеа над 60 еминентни професори и доктори од Клиничкиот центар Мајка Тереза од Скопје, како и докторите од здравствените установи од нашиот охридско – струшки регион – Охридската и Струшката болница, ЈЗУ Специјална болница за ортопедија и трауматологија “Св. Еразмо” – Охрид, ЈЗУ Специјализирана болница за превенција, лекување и рехабилитација на кардиоваскуларни заболувања “Свети Стефан” – Охрид како и доктори од многу приватни ординации од општина Охрид и општина Струга. Она што навистина загрижува е бројноста и различноста на ретките болести во овој регион. За жал, стигнавме и до информации за ултра ретки дијагнози како што се Peching syndrome, Tyrosinemia type 1, Rett syndrome, Gitelman syndrome…
Сепак надежта останува и понатаму да не води и искрено се надеваме дека ова истражување ќе ни помогне да изработиме база на податоци која ќе ни послужи за планирање на следните активности во интерес на лицата со ретки болести, на семејствата меѓусебно поврзување и размена на искуства. Она што не охрабрува и не мотивира да одиме напред е позитивниот ефект од кампањите кои ги водевме во изминатите години, свесни сме дека помогнавме и охрабривме многу пациенти и семејства да зборуваат јавно за својата болест, да соберат храброст да не исконтактираат, а секако добивме многу пофалби и од стручната медицинска фела која е сè поактивно вклучена во кампањите, а и ние упатуваме јавна благодарност затоа што без нивната помош и стручните совети немаше да успееме.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29-ти февруари, РЕДОК датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има РЕДОК број на денови.
Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008-ма година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година. Р.С. Македонија за прв пат се вклучи во одбележување на овој редок ден во 2012-та година. Кампањата за Денот на Ретки Болести е за пошироката јавност, за релевантните институции и секој е добредојден да се приклучи: пациенти и семејства, организации на пациенти, медицински професионалци, истражувачи, фармацевтска индустрија, јавни здравствени институции – колку повеќе не има, толку подобро. Денот на ретките болести е можност за учесниците да бидат дел од глобалниот повик кон креаторите на политики, истражувачите, компаниите и здравствените професионалци повеќе и поефикасно да ги вклучат пациентите во истражувањата за ретки болести.
ВКЛУЧЕТЕ СЕ!










{kind=link}
{kind=link}
{kind=link}
{kind=link}